Evo 2: A Groundbreaking Biological Foundation Model
The language of life is written in DNA, a four-letter code that orchestrates everything from bacterial metabolism to human cognition. For decades, scientists have sought to decode this language, but its complexity – spanning billions of years of evolution and trillions of nucleotides – has defied simple interpretation. Enter Evo 2, a groundbreaking AI model that reads, predicts, and even writes genomic sequences with unprecedented precision. Developed by researchers at the Arc Institute, Stanford, NVIDIA, and other institutions, Evo 2 represents a paradigm shift in computational biology. Let’s explore how this model works, what it can do, and why it matters.
Genomes: From Prokaryotes to Eukaryotes
Life exists at varying levels of complexity. Even some of the simplest organisms have a genetic code that specifies their structure and function. For example, most bacteria have a genome written onto a single chromosome that is several million base pairs long and circular, meaning it forms a loop and lacks telomeres. In contrast, eukaryotic organisms like humans have linear chromosomes with telomeres that require a more advanced biological mechanism to facilitate replication. Prokaryotes and eukaryotes also exhibit differences in gene structure. Prokaryotic genes are relatively simple whereas eukaryotic genes are, as the paper authors note, “characterized by extensive noncoding regions, alternative splicing patterns, and multiple layers of epigenomic control.” While previous models focused primarily on prokaryotic genomes, Evo 2 subsumes genomes from all domains of life, allowing it to operate at a variety of scales and complexities. Evo 2 promises to usher in a new era of synthetic biology, enabling the design of full-scale genomes and epigenomes with applications ranging from biotechnology to gene therapy.
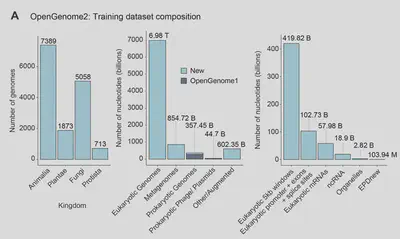
Data: The Fuel for Evo 2
The Architecture: StripedHyena and the Power of Scale
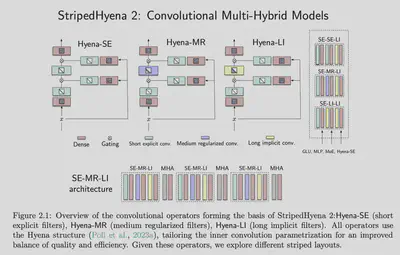
StripedHyena 2 is a convolutional, multi-hybrid architecture that uses three different types of input-dependent convolutional operators:
- Short-Length Hyena Operators: these are explicitly-parameterized and are designed to maximize hardware utilization while excelling at local, multi-token recall.
- Medium-Length Hyena Operators: these are regularized and designed for efficient modeling across hundreds of tokens.
- Long-Length Hyena Operators: these are implicit and intended to “aggregate information over the entire sequence.”
The innovations in StripedHyena 2 warrant a blog post of their own, but this is the gist of how the hybrid model composes operators designed to target sequence modeling at varying scale lengths. This multi-hybrid approach allows the model to capture both local patterns (e.g., codon triplets) and global dependencies (e.g., gene regulation across megabases).
Other Innovations:
In addition to the StripedHyena 2 architecture, Evo 2 boasts several other key innovations:
- 7B (trained on 2.4 trillion tokens) and 40B (trained on 9.3 trillion tokens) parameter model versions.
- Up to 1-Million-Token Context: Evo 2 processes sequences up to 1 million nucleotides long—enough to analyze entire human chromosomes or bacterial genomes in a single pass.
- Two-Phase Training:
- Pretraining: Focused on functional regions (genes, promoters) with 8,192-token context-length.
- Midtraining: Extended to 1M-token contexts using whole genomes, teaching the model long-range genomic syntax.
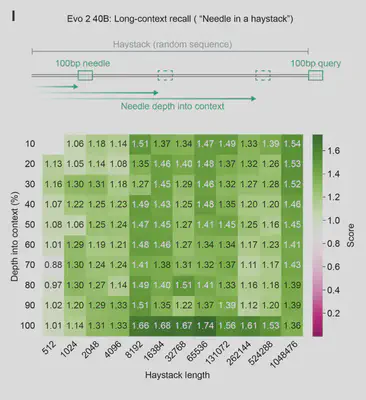
- Needle-in-Haystack Retrieval: The authors show on a synthetic long-context evaluation that the model can retrieve a 100 bp “needle” from a 1 million bp “haystack” with high accuracy in a random DNA sequence.
The result? A 40-billion-parameter behemoth that outperforms transformers in speed and accuracy, with an up to 1.3x speedup at 16K context length and up to 3x speedup at the 1M context length.
Capabilities: From Prediction to Design
Evo 2 isn’t just a passive observer—it’s a versatile tool for understanding and engineering biology. Previously, zero-shot mutational effect prediction was only possible for models trained exclusively on protein or prokaryotic sequences. Evo 2 extends this capability to a model that spans the entire central dogma (DNA, RNA, protein) and all three domains of life (prokaryota, archaea, eukaryota).
Zero-Shot Mutational Effect Prediction
Evo 2’s zero-shot prediction ability lets it score mutations without task-specific training:
- Pathogenicity: Outperforms specialized tools like AlphaMissense in classifying ClinVar variants, especially for noncoding and splice-altering mutations.
- Essentiality: Predicts gene knockout effects in bacteria and human lncRNAs with high accuracy, matching experimental screens.
- Evolutionary Constraints: Detects conserved elements like ribosome binding sites and stop codons across species, even distinguishing genetic codes (e.g., ciliate vs. standard stop signals).
Evo 2 Likelihoods Correlate with Function
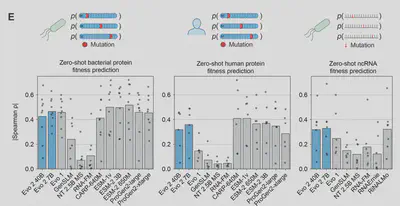
Beyond simple fitness predictions, Evo 2 is also capable of performing a more nuanced measurement of differential phenotypic consequences associated to variants based on their type and genomic location. Key points include:
- Coding Sequences: Evo 2 correctly predicts that non-synonymous mutations, frameshifts, and premature stop codons are more disruptive than synonymous mutations, reflecting their greater impact on protein function.
- Noncoding Sequences: Deletions in essential noncoding elements like tRNAs and rRNAs have larger effects than those in less critical regions (e.g., intergenic areas), consistent with their biological importance.
- Model Sensitivity: The larger 40B model shows heightened sensitivity to deletions in regulatory RNAs (e.g., miRNAs, snoRNAs), suggesting it captures finer regulatory details compared to the smaller 7B model.
- Benchmark Performance: Evo 2 outperforms other DNA language models in zero-shot tasks involving human noncoding regulatory sequences, demonstrating its ability to model complex, “fuzzy” genomic elements.
Human Clinical Variant Effect Prediction
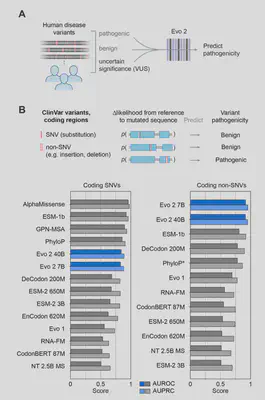
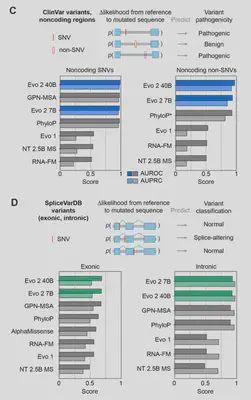
Further experiments were performed on BRCA1 and BRCA2 breast cancer variant datasets, but I’ll save those for a future post. In short, the researchers were able to leverage embeddings from Evo 2 model to perform more specialized variant effect predictions.
Peering Inside the Black Box: Mechanistic Interpretability
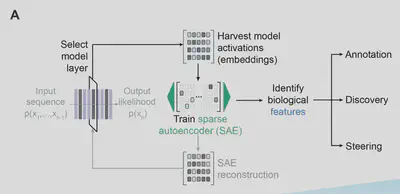
- Exon-Intron Architectures: The model autonomously learned features that activate preferentially on coding regions, enabling accurate genome annotation (even in woolly mammoths!).
- Genome Organization: Contrastive feature search revealed features corresponding to “open reading frames (ORFs), intergenic regions, tRNAs, and rRNAs, in the E. Coli genome.”
- Prophage Detection: A dedicated feature (f/19746) flagged phage-derived CRISPR spacers and annotated prophages in the E. coli genome.
- Protein Structure: There exist corresponding α-helices and β-sheets, linking DNA sequence to 3D protein secondary structures.
These insights validate Evo 2 as more than a mere pattern matcher—it’s a knowledge engine that rediscovers biology from first principles. If you’re curious about exploring some of Evo 2’s learned features yourself, the authors have made available an interactive tool that allows one to visualize Evo 2’s mechanistic interpretability across 104 genomes.
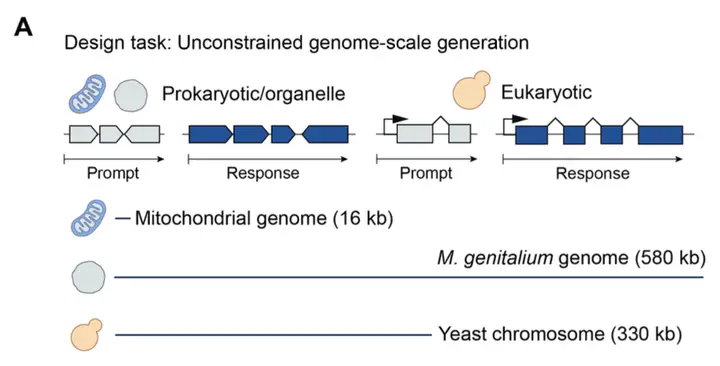
Autoregressive DNA Sequence Design at Genome Scale
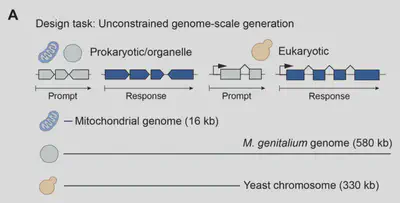
- Mitochondria: Generated 16,000-base mitochondrial genomes with correct numbers of tRNA, rRNA, and protein-coding genes, with protein complexes being predicted and validated using AlphaFold 3.
- Small Prokaryotic Genomes: Created M. genitalium-like genomes (~580,000 bases) where 70% of synthetic genes matched known protein domains as measured against the Pfam database. Generated proteins were assessed to be high-quality with respect to ESMFold metrics, secondary structure distribution, and protein sequence identity. Genomes were generated autoregressively, using the first 10.5 kb segment of the M. genitalium genome as a prompt.
- Yeast Chromosome and Eukaryotic Generation: Produced 330,000-base eukaryotic sequences with properly positioned promoters, introns, and tRNA clusters. Generated proteins were similar to those produced by natural yeast genes with regard to both sequence and structure.
Epigenomic Engineering via Inference-Time Search
Recent research has demonstrated the importance of the epigenome in regulating gene expression and cellular function. One component of epigenomic regulation involves the modification of the openness or compactness of chromatin, thereby controlling which parts of DNA are accessible to transcription factors and other proteins. The authors therefore sought to generate DNA sequences while specifying chromatin accessibility. Using an ensemble of Enformer and Borzoi models as “scoring functions” to predict how well chromatin accessibility matched a desired pattern, they optimized generations for specific epigenomic states—showcasing how AI can program biological function. The resulting method used beam search to score partial generations and match chromatin accessibility patterns to the desired state.
Using this method, the researchers were even able to encode Morse code messages
into the epigenomes generated by Evo 2.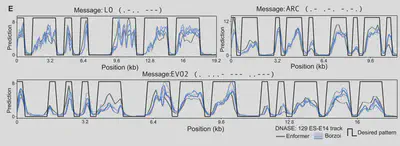
Open Science and Responsible Innovation
The team released Evo 2 openly, including:
- Model weights (7B and 40B)
- Training code and the OpenGenome2 dataset
- Interactive tools for generation and feature visualization
Safety measures were prioritized:
- Excluded Pathogens: Viral genomes (e.g., HIV, influenza, etc.) were omitted from training to curb misuse.
Conclusion
Evo 2 isn’t merely a tool for biologists—it’s a testament to what’s possible when machine learning meets life’s code. By reading genomes at scale, predicting variant effects, and writing synthetic DNA, it heralds a future where we program biology as easily as we code software. As the model’s capabilities grow, so too does our ability to heal, innovate, and explore the frontiers of life itself.
For researchers eager to experiment, Evo 2’s models and code are available on Hugging Face and GitHub.
Daniel McNeela
Machine Learning Researcher and Engineer
My research interests include patent and IP law, geometric deep learning, and computational drug discovery.